Meta’s newest AI determines proper protein folds 60 times faster
It’s ‘Mad Libs’ for O-Chem.
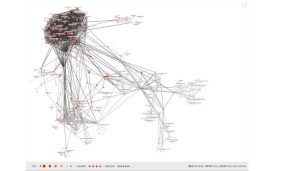
Life on Earth would not exist as we know it, if not for the protein molecules that enable critical processes from photosynthesis and enzymatic degradation to sight and our immune system. And like most facets of the natural world, humanity has only just begun to discover the multitudes of protein types that actually exist. But rather scour the most inhospitable parts of the planet in search of novel microorganisms that might have a new flavor of organic molecule, Meta researchers have developed a first-of-its-kind metagenomic database, the ESM Metagenomic Atlas, that could accelerate existing protein-folding AI performance by 60x.
Metagenomics is just coincidentally named. It is a relatively new, but very real, scientific discipline that studies “the structure and function of entire nucleotide sequences isolated and analyzed from all the organisms (typically microbes) in a bulk sample.” Often used to identify the bacterial communities living on our skin or in the soil, these techniques are similar in function to gas chromatography, wherein you’re trying to identify what’s present in a given sample system.
Similar databases have been launched by the NCBI, the European Bioinformatics Institute, and Joint Genome Institute, and have already cataloged billions of newly uncovered protein shapes. What Meta is bringing to the table is “a new protein-folding approach that harnesses large language models to create the first comprehensive view of the structures of proteins in a metagenomics database at the scale of hundreds of millions of proteins,” according to a Tuesday release from the company. The problem is that, while advances of genomics have revealed the sequences for slews of novel proteins, just knowing what those sequences are doesn’t actually tell us how they fit together into a functioning molecule and going figuring it out experimentally takes anywhere from a few months to a few years. Per molecule. Ain’t nobody got time for that.
“The ESM Metagenomic Atlas will enable scientists to search and analyze the structures of metagenomic proteins at the scale of hundreds of millions of proteins,” the Meta research team wrote on Tuesday. “This can help researchers to identify structures that have not been characterized before, search for distant evolutionary relationships, and discover new proteins that can be useful in medicine and other applications.”
Like languages, proteins are made up of their constituent atoms (think, words) which can all be smashed together as you wish but will only make a functional molecule (ie a coherent thought) if assembled in a specific order (a molecular sentence). Meta’s system drastically accelerates our capabilities to uncover organic chemistry’s syntax and grammar, however the analogy isn’t perfect. “A protein sequence describes the chemical structure of a molecule, which folds into a complex three-dimensional shape according to the laws of physics,” the team explained. “Protein sequences contain statistical patterns that convey information about the folded structure of the protein.”
Specifically, Meta’s Evolutionary Scale Modeling AI treats gene sequences like a Mad Libs for O-Chem using a self-supervised learning called masked language modeling. “We trained a language model on the sequences of millions of natural proteins,” the research team wrote. “With this approach, the model must correctly fill in the blanks in a passage of text, such as ‘To __ or not to __, that is the ________.’ We trained a language model to fill in the blanks in a protein sequence, like ‘GL_KKE_AHY_G’ across millions of diverse proteins.”
The resulting “protein language model” is named ESM-2 and operates across 15 billion parameters, making it the largest model of its kind to date. The “new structure prediction capability enabled us to predict sequences for the more than 600 million metagenomic proteins in the atlas in just two weeks on a cluster of approximately 2,000 GPUs.” So much for months and years.
(27)